Ceppi clinici di Listeria monocytogenes isolati nell'Umbria e nelle Marche nel biennio 2019-2020: come è cambiata la sorveglianza molecolare nell'era del Whole Genome Sequencing
Clinical strains of Listeria monocytogenes isolated in Umbria and Marche Regions (Italy) in 2019-2020: how molecular surveillance has changed in the age of Whole Genome Sequencing
Clinical strains of Listeria monocytogenes isolated in Umbria and Marche Regions (Italy) in 2019-2020: how molecular surveillance has changed in the age of Whole Genome Sequencing
[Ver. 1.0]
Fabrizia Guidi1, Antonietta Gattuso2, Marco Francesco Ortoffi2, Francesca Orecchioni3, Elisabetta Loggi4, Luciana Gironacci5, Antonella Repetto6, Gigliola Venditti7, Antonella Mencacci8, Patrizia Rinaldi9, Sauro Brodi10, Antongiulio Gallina11, Valerio Belfiori4, Maria Laura Baldoni12, Anna Duranti1, Elena Roccheggiani1, Stefania Scuota1, Viviana Bazzucchi1, Maira Napoleoni1, Cinzia Lorenzetti1, Barbara Palombo1, Adriano di Pasquale13, Cesare Cammà13, Francesco Pomilio14, Giuliana Blasi1
1 Istituto Zooprofilattico Sperimentale Umbria e Marche "Togo Rosati". Perugia.
2 Dipartimento di Sicurezza Alimentare, Nutrizione e Sanità Pubblica Veterinaria, Istituto Superiore di Sanità. Roma.
3 Azienda Ospedaliera Universitaria Ospedali Riuniti, Ancona
4 UOC Patologia Clinica, Fermo, ASUR-AV4.
5 UOC Patologia Clinica, Civitanova Marche, ASUR-AV3,
6 Azienda Ospedaliera di Perugia.
7 Ospedale Media Valle del Tevere, Todi (PG)
8 Dipartimento di Medicina e Chirurgia, Microbiologia e Microbiologia Clinica, Università degli studi di Perugia.
9 Ospedale di Narni (TR).
10 Ospedale città di Castello, USL Umbria 1 (PG).
11 Presidio Ospedaliero di Gubbio e Gualdo-Tadino, Gubbio (PG).
12 Ospedale San Giovanni Battista, Foligno (PG).
13 Centro di Referenza Nazionale per Sequenze Genomiche di Microrganismi Patogeni, Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise "G. Caporale", Teramo.
14 Laboratorio Nazionale di Riferimento per L.monocytogenes, Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise "G. Caporale", Teramo.
2 Dipartimento di Sicurezza Alimentare, Nutrizione e Sanità Pubblica Veterinaria, Istituto Superiore di Sanità. Roma.
3 Azienda Ospedaliera Universitaria Ospedali Riuniti, Ancona
4 UOC Patologia Clinica, Fermo, ASUR-AV4.
5 UOC Patologia Clinica, Civitanova Marche, ASUR-AV3,
6 Azienda Ospedaliera di Perugia.
7 Ospedale Media Valle del Tevere, Todi (PG)
8 Dipartimento di Medicina e Chirurgia, Microbiologia e Microbiologia Clinica, Università degli studi di Perugia.
9 Ospedale di Narni (TR).
10 Ospedale città di Castello, USL Umbria 1 (PG).
11 Presidio Ospedaliero di Gubbio e Gualdo-Tadino, Gubbio (PG).
12 Ospedale San Giovanni Battista, Foligno (PG).
13 Centro di Referenza Nazionale per Sequenze Genomiche di Microrganismi Patogeni, Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise "G. Caporale", Teramo.
14 Laboratorio Nazionale di Riferimento per L.monocytogenes, Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise "G. Caporale", Teramo.
Abstract. Listeria monocytogenes (Lm) is a major foodborne pathogen causing human listeriosis, the most severe zoonoses with the highest hospitalization and fatality rates (EFSA ECDC 2021). Invasive forms of the disease are particularly dangerous for the elderly, immunocompromised people, newborns and pregnant women, leading to sepsis, meningitis, encephalitis, abortion and stillbirth (Camargo et al. 2019). To Date Whole Genome Sequencing (WGS) allows an unprecedented subtyping resolution and is the best epidemiological surveillance tool in foodborne outbreak investigations and food monitoring programs. It completely replaced PFGE, for years considered the golden standard method for Lm typing. A joint ECDC-EFSA WGS database is being developed through the collaboration of Member States Competent Authorities for Lm. In Italy, the National Institute of Health (ISS) and the National Reference Laboratory for Lm (LNR-Lm) coordinate this activity. The Istituto Zooprofilattico Sperimentale dell'Umbria e delle Marche through the Food Control Laboratory of Fermo, acts as the Regional Reference Centre where all the isolates from clinical cases, food and environment are sent to be collected and typed using WGS and bioinformatic analysis in collaboration with the LNR-Lm and the ISS. The aim of this paper is to report the results coming from this activity in the years 2019 and 2020
Riassunto. Listeria monocytogenes (Lm) è l'agente eziologico della listeriosi, una patologia soggetta a notifica obbligatoria che si trasmette principalmente attraverso il consumo di alimenti contaminati. Le forme invasive della malattia colpiscono soprattutto soggetti appartenenti alle categorie a rischio, quali immunocompromessi, anziani e bambini, nei quali l'infezione provoca setticemia e meningite; anche nelle donne in gravidanza la listeriosi si manifesta in maniera severa, provocando aborto e natimortalità (Camargo et al., 2019). Rispetto alle altre zoonosi alimentari, la listeriosi, pur avendo una bassa prevalenza, presenta i tassi di ospedalizzazione e di mortalità più elevati (EFSA ECDC 2021). Il sequenziamento dell'intero genoma batterico (WGS) rappresenta ad oggi lo strumento con più alto potere discriminante nella tipizzazione degli isolati batterici. Questa tecnica ha ormai definitivamente sostituto la PFGE nella sorveglianza epidemiologica della listeriosi ed è ampiamente utilizzata nell'ambito delle indagini epidemiologiche, nell'identificazione degli outbreak e negli studi di source attribution. Con l'importanza crescente del WGS è nata la necessità di integrare l'EFSA-ECDC joint database anche con i dati ottenuti mediante questo tipo di analisi attraverso la collaborazione delle diverse Autorità Competenti. In Italia ad occuparsi di questa attività sono l'Istituto Superiore di Sanità e il Laboratorio Nazionale di Riferimento per Lm. L'Istituto Zooprofilattico Sperimentale dell'Umbria e delle Marche, tramite il Laboratorio Controllo Alimenti della sezione di Fermo, opera come Centro di Riferimento Regionale per Lm. Ad esso affluiscono tutti gli isolati clinici, alimentari ed ambientali provenienti dalle due regioni, per essere collezionati e tipizzati mediante WGS con il supporto dell'ISS e dell'LNR-Lm. Questo lavoro si propone di descrivere l'attività svolta in questo ambito negli anni 2019 e 2020.
Introduzione
La listeriosi è una patologia soggetta a notifica obbligatoria (D.M. 15/12/1990) che si trasmette principalmente attraverso il consumo di alimenti contaminati. L'agente eziologico responsabile è Listeria monocytogenes (Lm), bacillo Gram positivo ubiquitario e in grado di moltiplicarsi e/o sopravvivere in condizioni di stress quali l'ambiente refrigerato, l'acidità e l'elevata salinità (Maury et al., 2019).
Queste caratteristiche rendono Lm un problema piuttosto significativo nell'ambito della produzione alimentare, soprattutto per alcune categorie di alimenti quali i Ready To Eat, nei quali la contaminazione si verifica solitamente dopo la trasformazione e la crescita del patogeno è spesso supportata dalle caratteristiche fisico-chimiche del prodotto quali pH ed acqua libera (European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC), 2019).
La listeriosi umana si manifesta in due forme cliniche, la forma gastro intestinale, più frequente in soggetti immunocompetenti e la forma invasiva, che colpisce soprattutto soggetti appartenenti alle categorie a rischio, quali gli immunocompromessi, gli anziani e i bambini nei quali l'infezione provoca setticemia e meningite; anche per le donne in gravidanza la listeriosi si manifesta in maniera severa, provocando aborto e natimortalità (Camargo et al., 2019).
Rispetto ad altre zoonosi alimentari, la listeriosi, pur avendo una bassa prevalenza, presenta tassi di ospedalizzazione (92.1%) e di mortalità (17,6%) piuttosto elevati assumendo una particolare rilevanza sanitaria(European Food Safety Authority & European Centre for Disease Prevention and Control, 2021). Nella UE le infezioni da Lm sono oggetto di sorveglianza in sanità pubblica nell'ambito del programma Foodborne and Waterborne Diseases (FWD) dell'European Centre for Diseases Prevention and Control (ECDC) attraverso il sistema di sorveglianza europeo TESSy (The European Surveillance System) case-based (sorveglianza dei casi clinici di malattia) e TESSy isolate-based (sorveglianza molecolare sugli isolati di Lm) in conformità alla definizione di caso per la listeriosi (Decisione CE n. 945 del 22 Giugno 2018).
La sorveglianza si focalizza sulle forme invasive. Inoltre, al fine di garantire la sicurezza degli alimenti destinati all'uomo il Regolamento CE No 2073/2005 e s.m.i., stabilisce i criteri microbiologici di sicurezza alimentare per Lm negli alimenti.
Secondo i dati più recenti, nel 2019 i casi confermati di listeriosi invasiva verificatisi nei paesi dell'Unione Europea (UE) sono stati in tutto 2621.
Il tasso di notifica è stato di 0,46 per 100.000 abitanti, comparabile con il 2018 (0,47 casi per 100.000 abitanti) confermando la tendenza all'aumento dei casi di listeriosi negli ultimi 6 anni (2014-2019). Nello specifico, in Italia, nel 2019 sono stati segnalati 202 casi confermati di listeriosi invasiva nell'uomo, con un tasso di notifica di 0,33 casi per 100.000 abitanti. Il numero di focolai epidemici dovuti a Lm nel 2019 è risultato più alto del 50% rispetto al 2018 (European Food Safety Authority & European Centre for Disease Prevention and Control, 2021). La sorveglianza epidemiologica delle malattie a trasmissione alimentare non può prescindere dalla caratterizzazione molecolare degli isolati batterici e dall'individuazione delle correlazioni epidemiologiche tra i ceppi isolati da casi clinici e da alimenti contaminati. Nel caso di Lm, le tecniche di tipizzazione più utilizzate, per anni, sono state la sierotipizzazione, la Pulsed-Field Gel Electrophoresis (PFGE) e la Multi Locus Sequence Typing (MLST). La specie Lm presenta una grande eterogeneità genetica; al suo interno i ceppi possono essere raggruppati in 5 diversi sierogruppi, ulteriormente suddivisi su base fenotipica in 13 sierotipi, ed in vari MLST Clonal Complex (CCs).
Definire l'appartenenza degli isolati di Lm ad un determinato sierogruppo/sierotipo o con maggior potere discriminante, ad un CC (MLST), consente di individuare quelli più spesso associati a casi di listeriosi nell'uomo o più frequentemente isolati da specifiche categorie di alimenti, ottenendo informazioni importanti da un punto di vista epidemiologico e di source attribution.
Dei 13 diversi sierotipi di Lm conosciuti, i sierotipi 1/2a (sierogruppo IIa), 1/2b (sierogruppo IIb) e il 4b (sierogruppo IVb) provocano circa il 95% dei casi di listeriosi, mentre i sierotipi 1/2a, 1/2b e 1/2c (sierogruppo IIc), sono quelli più frequentemente isolati dagli alimenti. Il sierotipo 4b è quello che presenta la più forte associazione epidemiologica con casi di listeriosi umana ed è più spesso coinvolto in focolai epidemici (Chen et al., 2017; Lee et al., 2018; Montero et al., 2015).
Recentemente, Maury et al. (Maury et al., 2016, 2019) hanno riportato che all'interno dell'intera specie, il CC1, il CC2, il CC4 e il CC6, tutti appartenenti al sierotipo 4b, sono quelli significativamente più comuni tra gli isolati clinici rispetto a quanto ci si possa aspettare dalla loro prevalenza negli alimenti e sono pertanto definiti iper-virulenti. Al contrario il CC9 e il CC121, fortemente associati agli alimenti, sono definiti ipo-virulenti in quanto infettano principalmente individui altamente immunocompromessi (Maury et al., 2019).
Nel 2015 è stato istituito, a livello comunitario, un database per la raccolta dei dati di tipizzazione relativi ad isolati alimentari, ambientali ed umani di Salmonella, Listeria monocytogenes ed Escherichia coli produttori di Shiga-tossina (STEC). I dati di tipizzazione finora raccolti, sono stati i profili PFGE, per L. monocytogenes ed E. coli STEC e, solo per Salmonella, i profili di Multi-locus Variable Number Tandem Repeats Analysis (MLVA). Come evidenziato nella denominazione di "EFSA-ECDC joint database", i curatori di questo database, sono le due autorità comunitarie competenti del settore, la European Food Safety Authority (EFSA) e lo European Centre for Disease Prevention and Control (ECDC). La prima è responsabile e coordinatrice della raccolta dei dati riguardanti gli isolati alimentari ed ambientali mentre la seconda di quelli relativi agli isolati umani. L'implementazione di tale sistema ha permesso, negli anni, di individuare focolai di listeriosi anche multi-stato, grazie alla possibilità di confrontare i profili genetici di ceppi isolati in varie aree geografiche e in tempi diversi.
Questo ha consentito inoltre di rintracciare e ritirare dal commercio lotti di prodotti potenzialmente contaminati al fine di tutelare la salute del consumatore.
Negli ultimi anni stiamo assistendo all'utilizzo esponenziale del sequenziamento dell'intero genoma batterico (Whole genome sequencing -WGS) mediante la tecnologia del Next generation sequencing (NGS). La possibilità di ottenere in tempi rapidi la sequenza di interi genomi, fornisce uno strumento di caratterizzazione altamente discriminante per la sorveglianza epidemiologica, da impiegare nelle indagini relative ai casi di listeriosi, nell'identificazione di outbreak, negli studi di source attribution, nonché nelle attività di valutazione del rischio legato ai patogeni alimentari, consentendo una più mirata identificazione dei pericoli (European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC), 2019; Pettengill et al., 2014).
Sebbene vi sia ancora molto da fare in termini di standardizzazione delle piattaforme utilizzate, creazione di pipelines standard per l'analisi dei dati e di implementazione di database condivisi, a partire dal 2019, i metodi di tipizzazione basati sul WGS hanno sostituito definitivamente quelli tradizionali ed in particolare la PFGE, per anni considerata il gold-standard per la tipizzazione degli isolati batterici tra cui Lm. Attualmente, la metodica WGS più utilizzata per la tipizzazione di Lm è la core genome MLST (cgMLST). Lo schema utilizzato è quello implementato dall'Istituto Pasteur costituito da 1748 loci. Ad ogni modo, la determinazione di CC e sierogruppo, vengono ancora utilizzati come caratterizzazione iniziale ma si ricavano in silico (direttamente dalla sequenza dell'intero genoma) partendo dalle sequenze ottenute mediante NGS (European Centre for Disease Prevention and Control., 2016).
Il fattore limitante in quella che è ormai la nuova era della biologia molecolare, non è tanto la possibilità di acquistare piattaforme NGS e di generare sequenze "home made", dato il progressivo abbassamento dei costi dovuto alla velocità con cui questa tecnologia si sta evolvendo, bensì la capacità di assemblare, maneggiare ed analizzare grandi moli di dati attraverso sistemi standardizzati, ottenendo informazioni da utilizzare a fini diagnostici e di sorveglianza epidemiologica.
Il Ministero della Salute ha attivato, con Decreto del 30 maggio 2017, il "Centro di Referenza Nazionale per Sequenze Genomiche di microrganismi patogeni: banca dati e analisi di bioinformatica" (CRN GenPat) presso la sede centrale dell'Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise (IZSAM), il quale rappresenta una piattaforma nazionale per la raccolta e la conservazione delle sequenze genomiche di microrganismi patogeni, per l'esecuzione di analisi bioinformatiche, l'archiviazione e la condivisione dei risultati.
Il Centro inoltre fornisce un supporto tecnico-scientifico anche in termini di formazione. A livello Comunitario poi, diversi Centri di Eccellenza, tra i quali l'Istituto Pasteur di Parigi, hanno realizzato e reso disponibili, per alcuni microrganismi patogeni (L.monocytogenes, S.aureus, Salmonella spp., STEC ecc..) database online ( https://bigsdb.pasteur.fr/listeria/listeria.html) che, sulla base di algoritmi sviluppati per un'analisi rapida di interi genomi microbici, forniscono uno strumento per la comparazione filogenetica dei genomi e l'estrapolazione delle informazioni di interesse in essi contenute.
In questa stessa ottica, anche in Italia, il CNR GenPat, così come l'Istituto Superiore di Sanità (ISS), hanno messo a punto dei server pubblici (es. https://genpat.izs.it/, https://irida.iss.it/) per la raccolta e l'analisi dei genomi di microrganismi patogeni, fornendo uno strumento accessibile e di facile utilizzo per l'esecuzione di vari tipi di valutazioni (filogenetiche, presenza/assenza di geni di resistenza ad antibiotici/disinfettanti o di virulenza, ecc..).
L'auspicio è di rendere autonomi anche i Laboratori che non dispongono di una piattaforma NGS nell'analisi delle sequenze relative ai propri isolati, sequenze ottenute da Laboratori terzi. Con l'importanza crescente del WGS, inoltre, è nata la necessità di integrare l'EFSA-ECDC joint database anche con i dati ottenuti mediante questo tipo di analisi (European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC), 2019).
Le autorità competenti di ciascuno Stato Membro devono coordinarsi correttamente per la raccolta dei dati e dei risultati analitici da utilizzare per alimentare il database comunitario. In Italia, il flusso informativo e di dati, relativo ai ceppi di Lm e proveniente da ciascuna regione, viene gestito dal Laboratorio Nazionale di Riferimento per Lm (LNR) per quanto riguarda gli isolati alimentari ed ambientali e dall'ISS per quanto riguarda gli isolati clinici. Per l'Istituto Zooprofilattico Sperimentale dell'Umbria e delle Marche (IZSUM), il laboratorio di riferimento competente per territorio a cui afferiscono i ceppi di Lm isolati da pazienti, alimenti e ambienti di produzione, è il Laboratorio Controllo Alimenti di Fermo (LCA) che si occupa anche della gestione ed elaborazione dei relativi metadati. Negli ultimi anni inoltre il personale coinvolto in questa attività ha sviluppato competenze nell'ambito dell'analisi bioinformatica dei dati di sequenziamento degli isolati di Lm, di pari passo con l'evolversi dei metodi utilizzati come gold-standard per la tipizzazione batterica.
Scopo del presente lavoro è riportare i risultati dell'attività di sorveglianza epidemiologica molecolare della listeriosi svolta nelle regioni Marche e Umbria nel corso degli anni 2019 e 2020, mediante l'analisi bioinformatica delle sequenze di ceppi clinici all'interno delle piattaforme nazionali esistenti.
Materiali e Metodi
Raccolta dei ceppi e gestione delle relative informazioni epidemiologiche
Il sistema di sorveglianza della listeriosi nelle regioni di competenza dell'IZSUM, nato durante il focolaio di listeriosi verificatosi nelle Marche tra il 2015 e il 2016 e consolidatosi nel tempo, vede la collaborazione di tutte le Istituzioni pubbliche preposte al controllo delle Malattie Trasmesse da Alimenti (MTA). Nello specifico, il LCA di Fermo, coordina la raccolta dei ceppi di Lm isolati presso i diversi laboratori dell'IZSUM e dell'Agenzia Regionale per la Protezione dell'Ambiente Marche, da alimenti e tamponi ambientali prelevati dai Servizi dei Dipartimenti di Prevenzione dell'Azienda Sanitaria Unica Regionale. Al laboratorio di Fermo sono inoltre inviati i ceppi clinici di Lm isolati dai laboratori delle Aziende Sanitarie Ospedaliere delle regioni Umbria e Marche e dagli altri Laboratori Ospedalieri. Tutti i ceppi di Lm vengono tipizzati mediante Multiplex PCR per la determinazione del sierogruppo di appartenenza secondo quanto previsto dal protocollo fornito dal Laboratorio Europeo di Riferimento per Lm (EURL Lm) (Doumith et al., 2004; Guidi et al., 2021; Kérouanton et al., 2010). Tutti gli isolati vengono poi inviati al LNR per Lm, presso IZSAM, per essere caratterizzati mediante WGS. Il LCA di Fermo inserisce le informazioni relative ai ceppi inviati nel SEAP, il Sistema Informativo per la Sorveglianza epidemiologica agenti patogeni di origine alimentare, gestito dal LNR per conto del Ministero della Salute, ed elabora i risultati della sorveglianza. Una volta sequenziati, i genomi degli isolati sono disponibili, insieme con i relativi metadati, nella piattaforma genpat (https://genpat.izs.it/) implementata e gestita dal Centro di Referenza Nazionale per Sequenze Genomiche di microrganismi patogeni (CRN GenPat, IZSAM).
Tutti gli isolati clinici, insieme alle schede per la raccolta dei dati epidemiologici, sono inoltre inviati all'ISS che opera come Operational Contact Point microbiologico per Lm nel network FWD-ECDC in Italia. L'ISS a sua volta caratterizza i ceppi mediante WGS e carica i file di sequenziamento, insieme con i metadati riguardanti gli isolati, nella piattaforma integrata IRIDA (Integrated Rapid Infectious Disease Analysis) -ARIES (Advanced Research Infrastructure for Experimentation in genomicS).
Entrambe le piattaforme, richiedono un accesso riservato al personale competente individuato dalle Regioni.
Tipizzazione molecolare mediante WGS - Piattaforma bioinformatica genpat
Sequenziamento NGS
Il sequenziamento dei ceppi di Lm isolati da alimenti, superfici e pazienti affetti da listeriosi, è stato eseguito dal LNR e dal CRN GenPat mediante NGS, utilizzando la piattaforma Illumina (Illumina, San Diego, CA). Nello specifico, i ceppi sono stati sottoposti a estrazione del DNA utilizzando il Maxwell 16 tissue DNA purification kit (Promega Italia Srl, Milan, Italy), secondo le specifiche del produttore e la purezza degli estratti è stata poi valutata mediante lo strumento NanoDrop2000 (ThermoFisher Scientific, Wältham, MA). La preparazione delle librerie genomiche è stata realizzata Partendo da 1 ng di DNA mediante il kit Nextera XT DNA chemistry (Illumina, San Diego, CA). il sequenziamento è stato poi effettuato su piattaforma NextSeq500 Illumina con strategia PairedEnd reads 2x150 bp.
I dati di sequenza ottenuti sono stati analizzati all'interno della piattaforma genpat utilizzando una pipeline comprendente uno step di trimming con il software Trimmomatic v0.36 (Bolger et al., 2014) ed uno di controllo qualità delle reads con FastQC v0.11.5. Le coppie di reads sono state poi assemblate con metodo de novo utilizzando il software SPAdes v3.11.1. Il controllo di qualità per i genomi assemblati è stato realizzato con il software QUAST v.4.3 (Gurevich et al., 2013).
L'analisi bioinformatica delle sequenze con gli strumenti forniti dalla piattaforma è stata effettuata dal personale del LCA di Fermo dopo l'autenticazione con le proprie credenziali di accesso.
Multi Locus Sequence Typing (MLST) in silico
Lo schema di Multi Locus Sequence Typing (MLST) utilizzato per Lm è quello di Salcedo et al. (2003), basato sulla valutazione di un singolo polimorfismo (SNPs) di ciascuno di sette geni housekeeping presi in considerazione (abcZ, bglA, cat, dapE, dat, ldh, InkA). La variazione di un solo nucleotide determina l'assegnazione di uno specifico allele e la combinazione di questi sette alleli identifica uno specifico Sequence Type (ST) e un Clonal Complex (CC).
Il profilo MLST dei ceppi analizzati ed il relativo CC di appartenenza sono stati ottenuti in silico dalla piattaforma, utilizzando il Bacterial Isolate Genome Sequence Database (BIGSdb) dell'Istituto Pasteur di Parigi (https://bigsdb.pasteur.fr/listeria/).
Core Genome MLST
Per valutare con maggior potere discriminante le correlazioni genetiche esistenti tra i ceppi isolati, individuando eventuali cluster di infezione (cluster analysis) ed effettuando considerazioni di tipo epidemiologico, è stata utilizzata la core genome MLST (cgMLST). Come la MLST, questa tecnica si basa su un approccio definito gene-by-gene, che valuta le variazioni presenti nelle regioni codificanti del genoma batterico, ma ne costituisce un ampliamento, considerando non solo 7 geni ma migliaia. In particolare lo schema cgMLST utilizzato per l'analisi dei ceppi di Lm è quello definito dall'Istituto Pasteur, costituito da 1748 geni.
Per la definizione dei profili cgMLST è stato utilizzato chewBBACA (Comprehensive and Highly Efficient Workflow BSR-Based Allele Calling Algorithm), una pipeline bioinformatica completa (Silva et al., 2018) disponibile all'interno della piattaforma genpat. La visualizzazione delle correlazioni e delle distanze esistenti tra i profili allelici dei vari ceppi, è stata effettuata mediante un Minimun Spanning tree (MST) ricavato con il software GrapeTree (Zhou et al., 2018).
In accordo con quanto riportato da Moura et al. (Moura et al., 2017), ceppi con un numero di alleli di differenza inferiore o uguale a 7 (cut-off di similarità del 99,9%), sono stati come appartenenti allo stesso cluster. Piattaforma bioinformatica IRIDA-ARIES
Sequenziamento NGS
Per l'anno 2019 i ceppi clinici inviati all'ISS sono stati sottoposti a WGS dal Dipartimento di Sicurezza Alimentare, Nutrizione e Sanità Pubblica Veterinaria e le relative sequenze sono state caricate nella piattaforma IRIDA-ARIES.
Il DNA è stato estratto utilizzando il kit QuickExtractTM Bacterial DNA Extraction Kit (Lucigen Corporation, Middleton, WI), e per il sequenziamento è stata utilizzata la piattaforma Ion GeneStudio S5TM System (Thermo Fisher Scientific, Waltham, MA).
A partire dal 2020 poi, in accordo con i laboratori coinvolti nell'attività di WGS, le sequenze dei ceppi clinici prodotte dal LNR-Lm e dal CRN GenPat e disponibili sulla piattaforma genpat, sono state caricate, sotto forma di raw reads (file .fastq), direttamente nella piattaforma IRIDA-ARIES dal personale di Fermo. Una volta completato il caricamento dei file di sequenza, il sistema ha eseguito il controllo della qualità delle raw reads con il software FastQC. La pipeline automatica PHANtAsTiC 1.0 (Public Health Analysis of Nucleotides through Assembly, Typing and Clustering), ha poi effettuato in successione il trimming (Software Trimmomatic; Bolger et al., 2014), l'assemblaggio (INNUca; https://github.com/B-UMMI/INNUca) e la cluster analysis (cgMLST).
MLST in silico
Per la definizione del profilo MLST e del relativo ST, la piattaforma IRIDA-ARIES ha utilizzato il tool MentaLiST (Feijao et al., 2018).
Core Genome MLST
Per la definizione dei profili cgMLST secondo lo schema Pasteur, la piattaforma IRIDA-ARIES ha utilizzato il tool MentaLiST (Feijao et al., s.d.-b) incluso nell'Allele Observer Pipeline, con successivo calcolo della matrice delle distanze di Hamming tra ogni campione.
La visualizzazione delle correlazioni esistenti tra i profili allelici dei ceppi è stata eseguita attraverso un albero filogenetico Neighbor joining. Il cut-off di similarità utilizzato per la definizione di cluster è stato anche in questo caso quello riportato da Moura et al. (Moura et al., 2017) e corrispondente ad un massimo di 7 alleli di differenza.
Sorveglianza molecolare della Listeriosi basata sul WGS
Le correlazioni che i ceppi clinici isolati nelle regioni Umbria e Marche avevano tra di loro, sono state valutate sulla base dell'analisi di cgMLST. Utilizzando la piattaforma genpat, a fronte di isolati clinici che presentavano stesso sierogruppo e stesso CC, è stata lanciata l'analisi cgMLST al fine di verificare con maggior potere discriminante se essi facessero parte di uno stesso cluster di infezione. Con lo stesso tipo di analisi è stato inoltre possibile valutare eventuali correlazioni esistenti tra il genoma degli isolati clinici e quello di ceppi provenienti ad alimenti e superfici analizzati nel territorio umbro marchigiano nell'ambito del controllo ufficiale, anche in anni diversi rispetto a quelli in cui si erano verificati i casi di Listeriosi in oggetto.
Utilizzando la piattaforma IRIDA-ARIES invece, è stato possibile valutare le correlazioni che i ceppi clinici isolati in Umbria e nelle Marche avevano tra di loro e con isolati clinici di altre regioni. Per aumentare i livelli di sorveglianza, la piattaforma svolge automaticamente questo tipo di analisi ogni qualvolta la sequenza di un nuovo ceppo viene caricata e, nel caso in cui il sistema evidenzi l'esistenza di un cluster, invia un'e-mail di notifica a tutti gli utenti dei laboratori, delle Regioni coinvolte e al Ministero della Salute.
Caratterizzazione dei profili di antibiotico resistenza.
Le sequenze assemblate dei 37 genomi sono state scaricate in formato fasta dalla piattaforma genpat al fine di utilizzare il tool "Antibiotic Resistance" disponibile sul database BIGS-db (https://bigsdb.pasteur.fr/listeria/) per la valutazione dei profili di antibiotico resistenza. Questo tool utilizza uno schema predefinito, specifico per Lm, contenente 10 diversi geni di antibiotico resistenza: aacA4, aadC, aadE, aphA, cat_CHL, dfrD, ermB, fexA, fosX, lmo0919 (lin), lnuG, mphB, mprF, norB, penA, qnrB, str, sul, tetM, tetS (Moura et al., 2017).
Risultati e discussione
Nel periodo compreso tra il primo gennaio 2019 e il 31 dicembre 2020, i laboratori ospedalieri delle regioni Umbria e Marche hanno inviato al LCA di Fermo, 39 ceppi di Lm isolati da 35 casi di listeriosi umana (Tabelle 1 e 2). Di questi, 18 casi erano relativi a pazienti residenti nella regione Umbria, 15 a pazienti residenti nelle Marche e 2 a pazienti residenti in altre regioni e ricoverati in ospedali del territorio umbro marchigiano.
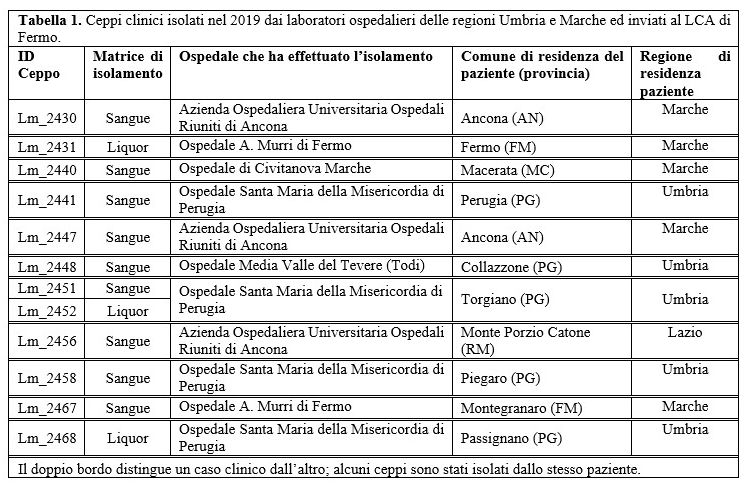
Table 1. Lm clinical strains isolated during 2019 in Umbria and Marche regions (Central Italy) and sent to LCA of Fermo by hospital laboratories.
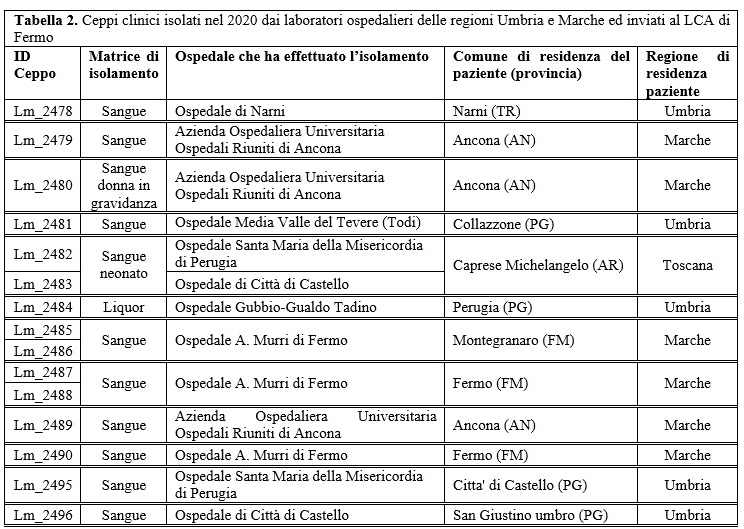
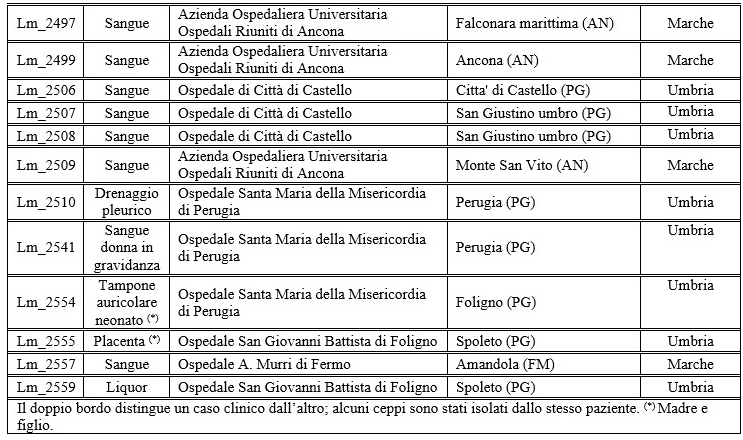
Table 2. Lm clinical strains isolated during 2020 in Umbria and Marche regions (Central Italy) and sent to LCA of Fermo by hospital laboratories.
Nella Tabella 3 è riportata la ripartizione dei casi di Listeriosi per regione e per anno.
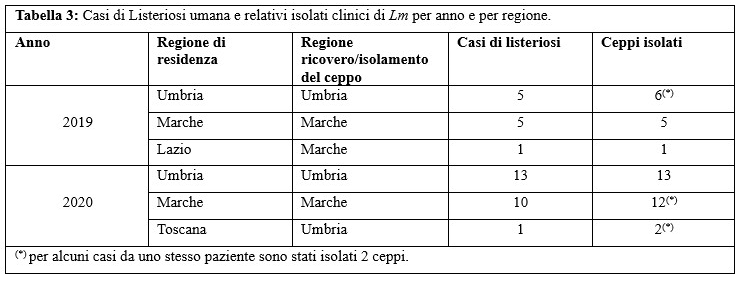
Table 3. Human listeriosis cases and related Lm strains by year and region of isolation
Il sierogruppo è stato determinato per tutti i 39 ceppi isolati: il 38% di essi era appartenente al sierogruppo IVb, il 33% al IIa, il 26% al IIb e il 3% al IIc. In Tabella 4 viene riportato il numero di ceppi di ogni sierogruppo, per regione e anno di isolamento.
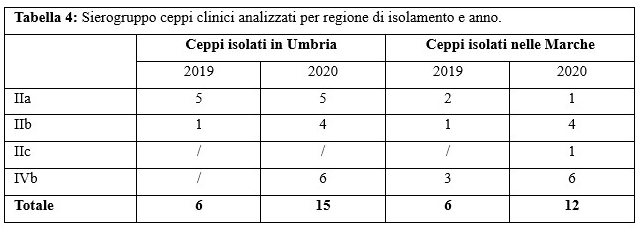
Table 4. Molecular serogrouping of Lm clinical strains indicated by year and region of isolation
Tutti i ceppi sono stati inviati al LNR-Lm e all'ISS insieme ai relativi metadati. I ceppi sottoposti a WGS sono stati in totale 37, corrispondenti a tutti gli isolati del 2019 e a 25 del 2020. Due sequenze, relative agli ultimi casi di Listeriosi verificatisi a fine 2020, non sono ancora disponibili. I 37 genomi ottenuti sono presenti sia su genpat che su IRIDA-ARIES.
Per quest'ultima piattaforma in particolare, 20 ceppi sono stati sequenziati ed inseriti dal personale dell'ISS mentre per i restanti 17, le sequenze, ottenute dal LNR-Lm e dal CRN GenPat, sono state scaricate da genpat dal personale del LCA di Fermo ed inserite in IRIDA-ARIES.
Attraverso l'analisi MLST sono stati identificati 14 diversi CC: 11 ceppi appartenevano al CC1 (29,7%), 5 al CC5 (13,5%), 4 al CC3 (10,8%), 3 al CC2 (8,1%), 3 al CC155 (8,1%), 2 al CC8 (5,4%), 2 al CC37 (5,4%) e un solo ceppo (2,7%) è stato isolato per il CC4, il CC7, il CC14, il CC19, il CC29, il CC101 e il CC451. Nella tabella 5 viene riportata la distribuzione dei CC per regione e anno di isolamento.
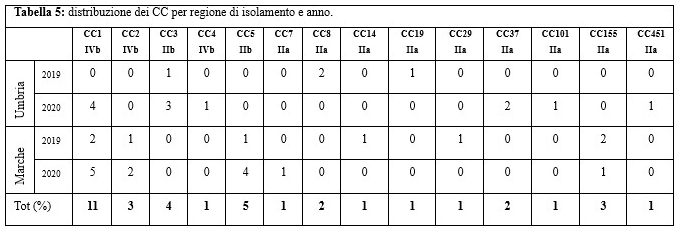
Table 5. Lm CCs distribution by year and region of isolation
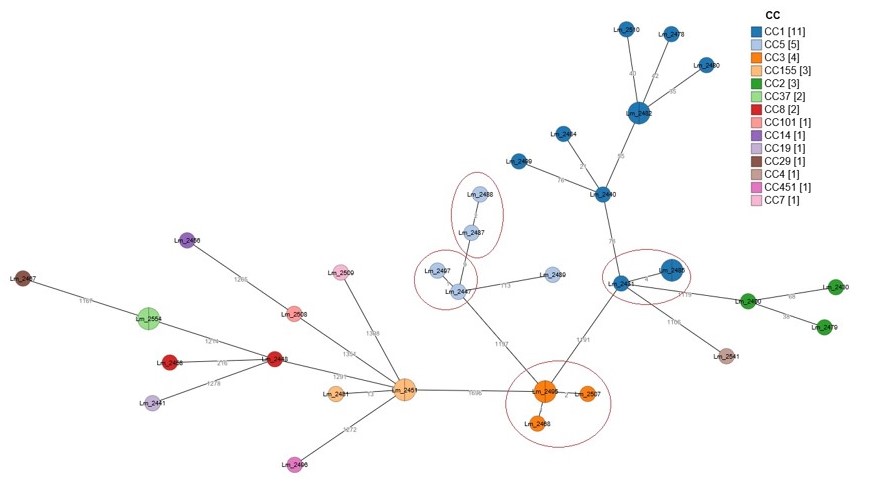
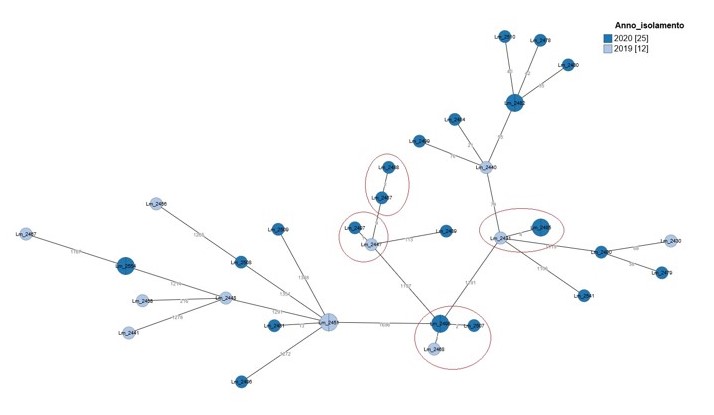
Le figure 1 e 2 mostrano due diverse rappresentazioni dell'albero MSTree ottenuto dall'analisi cgMLST con la piattaforma genpat e sul quale sono rappresentate le correlazioni genetiche tra tutti gli isolati clinici del periodo in oggetto.
cliccare per ingrandire l'immagine
Figura 1. Piattaforma genpat: analisi cgMLST dei ceppi clinici di Lm isolati nel periodo 2019-2020 nelle Regioni Umbria e Marche. I nodi dell'albero rappresentano i ceppi colorati per CC di appartenenza e i numeri riportati sui rami, costituiscono le distanze esistenti tra di essi in termini di differenze alleliche. I nodi Lm_2554, Lm_2451, Lm_2495, Lm_2485 e Lm_2482, contengono un altro isolato oltre a quello indicato, nello specifico il Lm_2555, Lm_2452, Lm_2506, Lm_2486 e Lm_2483 rispettivamente.
Figure 1. Genpat bioinformatic platform: cgMLST analysis of Lm strains isolated during 2019-2020 from clinical cases of human listeriosis occurred in Umbria and Marche Regions (Central Italy). The nodes of the Minimum Spanning tree represent the strains colored by CC to which they belong. Number values between adjacent nodes indicate the number of allelic differences between nodes. Note: the Lm_2554, Lm_2451, Lm_2495, Lm_2485 and Lm_2482 nodes also include Lm_2555, Lm_2452, Lm_2506, Lm_2486 and Lm_2483 respectively. |
cliccare per ingrandire l'immagine
Figura 2. Piattaforma genpat: analisi cgMLST dei ceppi clinici di Lm isolati nel periodo 2019-2020 nelle regioni Umbria e Marche. I nodi dell'albero rappresentano i ceppi colorati per anno d'isolamento e i numeri riportati sui rami, costituiscono le distanze esistenti tra di essi in termini di differenze alleliche. I nodi Lm_2554, Lm_2451, Lm_2495, Lm_2485 e Lm_2482, contengono un altro isolato oltre a quello indicato, nello specifico Lm_2555, Lm_2452, Lm_2506, Lm_2486 e Lm_2483 rispettivamente.
Figure 2. Genpat bioinformatic platform: cgMLST analysis of Lm strains isolated during 2019-2020 from clinical cases of human listeriosis occurred in Umbria and Marche Regions (Central Italy). The nodes of the Minimum Spanning tree represent the strains colored by year of isolation. Number values between adjacent nodes indicate the number of allelic differences between nodes. Note: the Lm_2554, Lm_2451, Lm_2495, Lm_2485 and Lm_2482 nodes also include Lm_2555, Lm_2452, Lm_2506, Lm_2486 and Lm_2483 respectively |
In accordo con la definizione di cluster data da Mora et al. (2017), l'analisi cgMLST eseguita su genpat, ha evidenziato quattro diversi cluster genetici. Gli isolati provenienti dallo stesso paziente si collocavano tutti nello stesso nodo e pertanto sono stati considerati come stesso ceppo (Figure 1 e 2). Due cluster appartenevano al CC5 (Lm_2447, Lm_2497 e Lm_2487, Lm_2488), uno al CC3 (Lm_2468, Lm_2495, Lm_2506 e Lm_2507) e l'altro al CC1 (Lm_2431 e Lm_2485-2486) (Figura 1). Tre dei cluster individuati, presentavano all'interno sia isolati del 2019 che isolati del 2020 (Figura 2). Utilizzando il genpat, contenente anche le sequenze di ceppi isolati da alimenti e ambienti di produzione nelle regioni in oggetto, è stato inoltre possibile valutare le correlazioni esistenti tra questi e tutti i ceppi clinici sopra descritti. In particolare, è stata fatta una selezione dei ceppi alimentari e ambientali isolati tra il 2018 e il 2020 per i quali erano disponibili le sequenze nella piattaforma, scegliendone solitamente almeno uno per campione. È stata poi avviata la pipeline per l'analisi di cgMLST includendo insieme a questi isolati, tutti i clinici del biennio 2019-2020. L'albero MSTree così ottenuto (Figura 3), ha evidenziato una stretta correlazione tra il ceppo Lm_2481, associato a un caso di Listeriosi verificatosi in Umbria nel 2020, e un ceppo proveniente da un campione di salmone affumicato prelevato nelle Marche nel 2018. Anche il ceppo Lm_2458, isolato da un paziente umbro nel 2019, è risultato molto vicino a due Lm isolate da tranci di tonno destinati alla produzione di sushi e prelevati nella regione Marche sempre nel 2019.
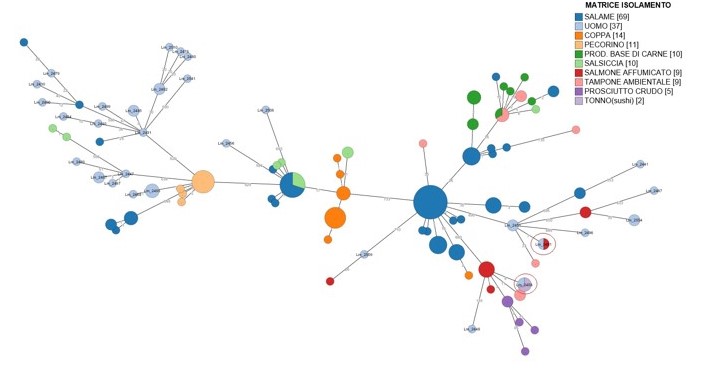
cliccare per ingrandire l'immagine
Figura 3. Figura 3. Piattaforma genpat: analisi cgMLST dei ceppi di Lm isolati da casi di Listeriosi umana tra il 2019 e il 2020 e da alimenti e superfici analizzati dal 2018 al 2020. L'identificativo è riportato esclusivamente per gli isolati clinici
Figure 3. Genpat bioinformatic platform: cgMLST analysis of Lm strains isolated from clinical cases of human listeriosis occurred in Umbria and Marche Regions (Central Italy) during 2019-2020 and from food and environmental samples tested between 2018 and 2019. Only for clinical isolates the id is reported |
In IRIDA-ARIES, 24 dei 37 ceppi clinici, rientravano in cluster genetici individuati dall'analisi di cgMLST svolta in automatico dal sistema su tutti gli isolati depositati nella piattaforma nazionale. In tutto, i cluster che presentavano all'interno ceppi isolati nell'Umbria e nelle Marche tra il 2019 e il 2020 erano 13, 10 dei quali includevano anche ceppi da casi di Listeriosi umana verificatisi in altre regioni (Tabella 6).
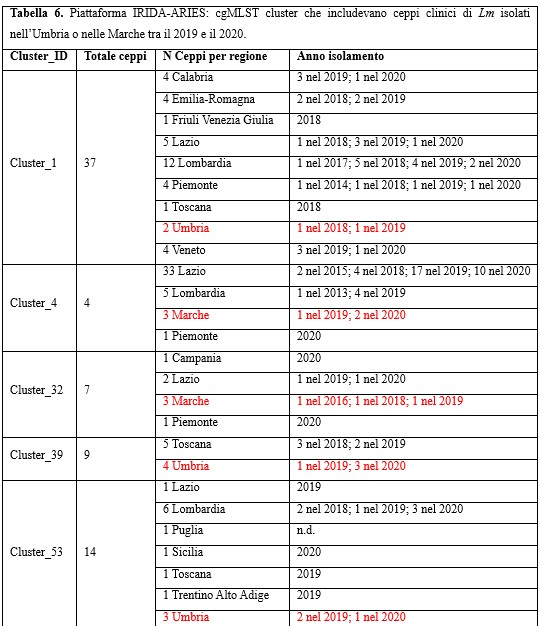
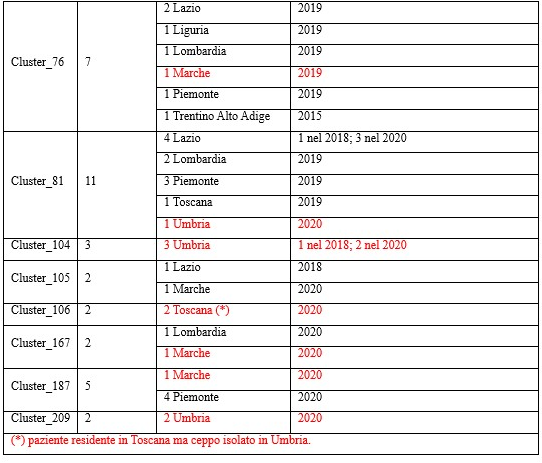
Table 6. IRIDA-ARIES bioinformatic platform: cgMLST clusters including Lm clinical strains isolated in Umbria and Marche Regions (Central Italy) during 2019-2020
Tra questi, per il Cluster_39, composto da 9 ceppi isolati tra il 2018 e il 2020 di cui 4 in Umbria (riferibili a 4 casi) e 5 in Toscana (riferibili a 4 casi), sono stati effettuati ulteriori approfondimenti, dal momento che i casi riguardavano due regioni confinanti e 6 di essi si erano verificati in un periodo di tempo piuttosto ristretto (dicembre 2019 a luglio 2020).
I ceppi umbri coinvolti, appartenenti come gli altri al sierogruppo IIb e al CC3, erano il Lm_2468, Lm_2495, Lm_2506 e Lm_2507 e rientravano tutti nel medesimo cluster anche nel sistema genpat (Figura 2).
A seguito delle e-mail di alert, ricevute dal sistema IRIDA-ARIES, sono stati presi i contatti con le Autorità Competenti preposte delle due regioni coinvolte ed è stato così possibile condividere le informazioni necessarie per approfondire e valutare l'eventuale nesso epidemiologico esistente tra i diversi casi.
Nel rispetto della privacy, tutte le informazioni anagrafiche e cliniche dei pazienti umbri e toscani associati al Cluster_39 sono riportate rispettivamente nelle Tabelle 7 e 8.
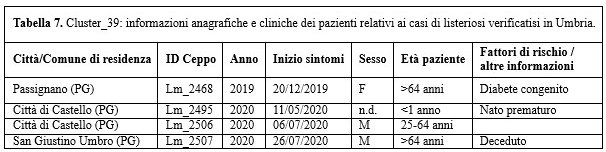
Table 7. Cluster_39: Personal and clinical information of patients related to listeriosis cases occurred in Umbria Region (Central Italy)
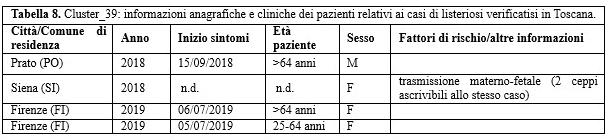
Table 8. Cluster_39: personal and clinical information of patients related to listeriosis cases occurred in Tuscany Region (Central Italy)
Il LCA di Fermo ha condiviso con le Autorità Regionali Competenti le indagini epidemiologiche svolte e, sulla base delle informazioni acquisite, è emerso che tutti i pazienti "intervistati" avevano consumato varie tipologie di prodotti a base di carne, in particolare affettati, acquistati presso la Grande Distribuzione. In nessuno dei casi investigati, è stato però possibile individuare un nesso diretto di casualità né un alimento comune ai diversi casi.
Per quanto riguarda l'analisi dei profili di resistenza agli antibiotici, tutti i genomi, analizzati interrogando il database BIGS-db, presentavano i geni fosX, norB e sul rispettivamente coinvolti nella resistenza a fosfomicina, fluorochinoloni e sulfonamidi (Leclercq et al., 2018; Haubert et al., 2018). In 35 ceppi è stato individuato anche il gene lin (lmo0919) per la resistenza alla lincomicina. Solo in due ceppi è stato rilevato il gene mprF, il quale conferisce una minore suscettibilità nei confronti dei peptidi cationici antimicrobici e degli antibiotici cationici (vancomicina, daptomicina).
Discussioni e Conclusioni
Nelle regioni Umbria e Marche l'attività di sorveglianza molecolare della listeriosi umana, svolta dal LCA di Fermo nel biennio 2019-2020, ha riguardato 39 ceppi di Lm provenienti da 35 casi di malattia. Nel 2020, il numero di casi notificati per i quali il LCA di Fermo ha ricevuto isolati (24), è stato quasi il doppio rispetto al 2019 (11), nonostante la situazione di forte stress del sistema sanitario, travolto dalla pandemia da SARS-CoV-2. Questo dimostra come il sistema implementato, che prevede il coinvolgimento di tutte le Istituzioni pubbliche preposte al controllo delle Malattie Trasmesse da Alimenti (MTA), sia ormai consolidato nelle regioni di competenza dell'IZSUM. Il coordinamento di tutte le attività e delle diverse competenze nell'ambito della sorveglianza epidemiologica e molecolare, ha consentito di gestire un elevato numero di ceppi batterici, con tempistiche ottimali e una buona suddivisione del lavoro.
Grazie allo sviluppo di competenze nell'ambito dell'analisi bioinformatica dei dati di sequenziamento, da parte del personale coinvolto, e all'implementazione di piattaforme nazionali per lo stoccaggio e l'analisi delle sequenze genomiche degli isolati, è stato possibile approfondire notevolmente la tipizzazione dei ceppi di Lm. Quest'approccio si rivela vincente nello studio della popolazione microbica associata ad un determinato territorio. Nell'ambito della sorveglianza epidemiologica, infatti, i metodi di tipizzazione molecolare possono essere utilizzati per monitorare la diffusione geografica e i cambiamenti prevalenti di cloni epidemici o endemici con l'obiettivo di valutare strategie preventive a lungo termine e di controllare di infezioni emergenti e riemergenti.
Nell'ambito dei ceppi clinici isolati nel periodo in oggetto, i sierogruppi maggiormente rappresentati erano il IVb, il IIa e il IIb. Questo risultato è in linea con quanto riportato in letteratura. È noto, infatti, che dei 13 diversi sierotipi di Lm conosciuti, l'1/2a (sierogruppo IIa), l'1/2b (IIb) e il 4b (IVb) provocano circa il 95% dei casi di listeriosi (Chen et al., 2017; Kuch et al., 2018; Wang et al., 2018) con il 4b che risulta più frequentemente associato a focolai epidemici (Montero et al., 2015).
L'analisi MLST in silico ha individuato 14 diversi CC mostrando un'elevata eterogeneità della popolazione batterica studiata, dal punto di vista genetico. Tra i CC rilevati ve ne erano alcuni definiti iper-virulenti in numerosi studi, in virtù di un'elevata frequenza di isolamento da casi di listeriosi e di una maggiore invasività riscontrata in vitro, associata alla presenza di specifici fattori di virulenza (Maury et al., 2016, 2019). In particolare, il CC1 è stato il CC più frequentemente isolato in assoluto, a differenza del CC2 del CC4 che invece erano scarsamente rappresentati. Questo CC è stato rilevato nelle Marche sia nel 2019 che nel 2020, mentre in Umbria compare solo nel 2020. Dopo il CC1, anche il CC5 e il CC3, entrambi responsabili di recenti focolai verificatisi in altri stati (Maury et al., 2016; Zhang et al., 2019), sono risultati particolarmente diffusi. L'isolamento di CC non particolarmente virulenti da pazienti affetti di listeriosi, sebbene meno frequente, dimostra come nonostante vi siano delle differenze in termini di invasività tra i vari cloni, qualsiasi ceppo di Lm sia verosimilmente in grado di provocare una forma invasiva di malattia come risultato di una complessa interazione tra il patogeno, l'alimento e le condizioni immunitarie dell'ospite.
L'analisi del profilo genetico di antibiotico-resistenza, è stata eseguita al fine di valutare la circolazione di ceppi aventi nel proprio genoma, geni implicati nella resistenza verso molecole utilizzate nelle terapie comunemente somministrate in caso di listeriosi. A riguardo, le molecole di prima scelta nel trattamento delle forme invasive includono i β-lattamici da soli o in associazione con un aminoglicoside; nello specifico, le combinazioni più somministrate sono gentamicina e penicillina o gentamicina e ampicillina (Khademi and Sahebkar, 2019). Anche altri antibiotici sono spesso utilizzati come seconda scelta, ma ultimamente, sono state riportate in Lm, resistenze nei confronti di alcune di queste molecole, e in particolare, nei confronti di fluorochinoloni, macrolidi, sulfonamidi e tetracicline (Khademi and Sahebkar, 2019). Per questo motivo, sebbene qualsiasi trattamento venga somministrato dopo aver eseguito un antibiogramma sul ceppo isolato dal paziente, è comunque importante, nell'ambito della sorveglianza, valutare la presenza dei geni di resistenza nei ceppi in circolazione. Dall'analisi dei profili di resistenza dei ceppi oggetto di studio, è emerso che tutti presentavano i geni fosX, norB e sul. Questo risultato è coerente con quanto riportato in letteratura. È noto, infatti, che Lm sia intrinsecamente resistente alla fosfomicina, per via della mancanza di un sistema di trasporto attraverso la membrana. La notevole diffusione del gene fosX riportata in questa specie costituisce un altro meccanismo alla base di questa resistenza ed è in linea con i quanto osservato nei ceppi analizzati (Lüth et al., 2020; Mota et al., 2020). Anche la presenza dei geni norB e sul è spesso riportata in Lm. Il primo conferisce resistenza nei confronti dei chinoloni quali norofloxacina, ciprofloxacina o sparfloxacina grazie al suo meccanismo di pompa di efflusso, mentre il secondo è associato a resistenza nei confronti dei sulfonamidi (Haubert et al., 2018; Kuch et al., 2018; Mota et al., 2020). Lm è considerata naturalmente resistente anche nei confronti della lincomicina e la presenza del gene lin è spesso riportata in letteratura (Lüth et al., 2020). I risultati ottenuti sui ceppi in oggetto sono pertanto in accordo con studi precedenti. Solo 2 isolati presentavano il gene mprf che conferisce una minore suscettibilità nei confronti dei peptidi cationici antimicrobici e degli antibiotici cationici (vancomicina, daptomicina) normalmente non utilizzati nel trattamento della listeriois(Bao et al., 2012; Thitiananpakorn et al., 2020).
Il maggior potere discriminante fornito dal WGS, insieme con la possibilità di valutare le correlazioni genetiche esistenti tra i ceppi isolati da casi di listeriosi, da alimenti e superfici di lavorazione sul territorio di competenza (piattaforma genpat) e tra ceppi clinici provenienti da diverse regioni del Paese (piattaforma IRIDA-ARIES), rappresenta un potente strumento per il riconoscimento e lo studio dei focolai di infezione e per l'individuazione degli alimenti responsabili nell'ambito indagini epidemiologiche relative anche a casi sporadici. Tuttavia, la rilevazione, all'interno di uno stesso cluster, di ceppi di Lm clinici ed alimentari, pur indicando una stretta correlazione genetica tra di essi, non è, da sola, sufficiente ad identificare un alimento come responsabile di infezione.
Il supporto di un nesso di casualità è infatti indispensabile. Nel corso dei due anni di sorveglianza descritti, sono stati identificati alcuni isolati clinici strettamente correlati con ceppi isolati da campioni alimentari; tuttavia questi dati analitici non hanno avuto riscontro dal punto di vista epidemiologico. Lo stesso vale nel caso di stretta correlazione genetica tra ceppi clinici associati a casi diversi di listeriosi; la possibilità che si tratti di un focolaio deve essere sostenuta dall'individuazione di un nesso epidemico.
Nel caso del Cluster_39, infatti, malgrado la correlazione molecolare tra ceppi clinici diversi e la stretta vicinanza geografica e temporale dei relativi casi di listeriosi abbiano suggerito la necessità di un approfondimento epidemiologico, non è stato possibile identificare un nesso causale quale l'esposizione dei pazienti a una fonte comune d'infezione.
La possibilità di avere a disposizione l'intera sequenza genomica di ceppi isolati da casi clinici, da alimenti e superfici di lavorazione, disponendo al contempo di diverse soluzioni per l'analisi bioinformatica, hanno incrementato notevolmente il potere discriminante della tipizzazione. La presenza di piattaforme nazionali, accessibili al personale competente, per la raccolta, la conservazione e l'analisi delle sequenze, ha rappresentato negli ultimi anni una svolta epocale nella sorveglianza molecolare della listeriosi e di altre zoonosi di particolare interesse, sebbene vi sia ancora molto da fare in termini di interconnessione tra le piattaforme bioinformatiche utilizzate, creazione di pipelines standard per l'analisi dei dati ed implementazione di database condivisi.
La produzione di una grande mole di dati molecolari in tempi brevi, genera l'opportunità di analizzare il genoma degli isolati in maniera immediata e sistematica, al fine di individuare eventuali correlazioni significative in tempi utili per un intervento efficace. Malgrado il ruolo basilare rivestito dalla sorveglianza molecolare nell'evidenziare possibili correlazioni tra casi, è necessario sensibilizzare il territorio in merito all'importanza fondamentale di svolgere approfondimenti epidemiologici in maniera tempestiva e secondo procedute condivise, coinvolgendo diverse competenze e con un approccio multidisciplinare.
La collaborazione con il LNR e l'ISS, i due enti di riferimento per Lm, ed il supporto tecnico-scientifico da essi fornito sia nell'ambito del sequenziamento degli isolati che nella formazione del personale in merito all'analisi delle sequenze, hanno permesso al LCA di Fermo di allinearsi a quella che è ormai la nuova era della tipizzazione microbica e della sorveglianza epidemiologica.
Ringraziamenti
Si ringrazia tutto il personale tecnico dei laboratori che ha supportato l'isolamento, la collezione e la caratterizzazione dei ceppi di L. monocytogenes negli anni oggetto del lavoro.
Si ringrazia tutto il personale tecnico dei laboratori che ha supportato l'isolamento, la collezione e la caratterizzazione dei ceppi di L. monocytogenes negli anni oggetto del lavoro.
Bibliografia
Bao, Y., Sakinc, T., Laverde, D., Wobser, D., Benachour, A., Theilacker, C., Hartke, A., Huebner, J. (2012). Role of mprF1 and mprF2 in the Pathogenicity of Enterococcus faecalis. PLoS ONE, 7(6), e38458. https://doi.org/10.1371/journal.pone.0038458
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114-2120. https://doi.org/10.1093/bioinformatics/btu170
Camargo, A. C., Moura, A., Avillan, J., Herman, N., McFarland, A. P., Sreevatsan, S., Call, D. R., Woodward, J. J., Lecuit, M., Nero, L. A. (2019). Whole-genome sequencing reveals Listeria monocytogenes diversity and allows identification of long term persistent strains in Brazil. Environmental Microbiology, 21(12), 4478-4487. https://doi.org/10.1111/1462-2920.14726
Chen, Y., Luo, Y., Carleton, H., Timme, R., Melka, D., Muruvanda, T., Wang, C., Kastanis, G., Katz, L. S., Turner, L., Fritzinger, A., Moore, T., Stones, R., Blankenship, J., Salter, M., Parish, M., Hammack, T. S., Evans, P. S., Tarr, C. L., � Brown, E. W. (2017). Whole Genome and Core Genome Multilocus Sequence Typing and Single Nucleotide Polymorphism Analyses of Listeria monocytogenes Isolates Associated with an Outbreak Linked to Cheese, United States, 2013. Applied and Environmental Microbiology, 83(15), e00633-17, e00633-17. https://doi.org/10.1128/AEM.00633-17
Doumith, M., Buchrieser, C., Glaser, P., Jacquet, C., Martin, P. (2004). Differentiation of the Major Listeria monocytogenes Serovars by Multiplex PCR. Journal of Clinical Microbiology, 42(8), 3819-3822. https://doi.org/10.1128/JCM.42.8.3819-3822.2004
European Centre for Disease Prevention and Control. (2016). Expert opinion on whole genome sequencing for public health surveillance: Strategy to harness whole genome sequencing to strengthen EU outbreak investigations and public health surveillance. Publications Office. https://data.europa.eu/doi/10.2900/12442
European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC). (2019). The European Union One Health 2018 Zoonoses Report. EFSA Journal, 17(12). https://doi.org/10.2903/j.efsa.2019.5926
European Food Safety Authority, European Centre for Disease Prevention and Control. (2021). The European Union One Health 2019 Zoonoses Report. EFSA Journal, 19(2). https://doi.org/10.2903/j.efsa.2021.6406
Feijao, P., Yao, H.-T., Fornika, D., Gardy, J., Hsiao, W., Chauve, C., Chindelevitch, L. (s.d.-a). MentaLiST - A fast MLST caller for large MLST schemes. Microbial Genomics, 8.
Feijao, P., Yao, H.-T., Fornika, D., Gardy, J., Hsiao, W., Chauve, C., Chindelevitch, L. (s.d.-b). MentaLiST - A fast MLST caller for large wgMLST schemes. 15.
Guidi, F., Orsini, M., Chiaverini, A., Torresi, M., Centorame, P., Acciari, V. A., Salini, R., Palombo, B., Brandi, G., Amagliani, G., Schiavano, G. F., Massacci, F. R., Fisichella, S., Domenico, M. D., Ancora, M., Pasquale, A. D., Duranti, A., Cammà, C., Pomilio, F., Blasi, G. (2021). Hypo- and Hyper-Virulent Listeria monocytogenes Clones Persisting in Two Different Food Processing Plants of Central Italy. Microorganisms, 9(2), 376. https://doi.org/10.3390/microorganisms9020376
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics, 29(8), 1072-1075. https://doi.org/10.1093/bioinformatics/btt086
Haubert, L., Kremer, F. S., da Silva, W. P. (2018). Whole-genome sequencing identification of a multidrug-resistant Listeria monocytogenes serotype 1/2a isolated from fresh mixed sausage in southern Brazil. Infection, Genetics and Evolution, 65, 127-130. https://doi.org/10.1016/j.meegid.2018.07.028
Kérouanton, A., Marault, M., Petit, L., Grout, J., Dao, T. T., Brisabois, A. (2010). Evaluation of a multiplex PCR assay as an alternative method for Listeria monocytogenes serotyping. Journal of Microbiological Methods, 80(2), 134-137. https://doi.org/10.1016/j.mimet.2009.11.008
Khademi, F., Sahebkar, A. (2019). A systematic review and meta-analysis on the prevalence of antibiotic-resistant Listeria species in food, animal and human specimens in Iran. Journal of Food Science and Technology, 56(12), 5167-5183. https://doi.org/10.1007/s13197-019-04040-w
Kuch, A., Goc, A., Belkiewicz, K., Filipello, V., Ronkiewicz, P., Golebiewska, A., Wróbel, I., Kiedrowska, M., Wasko, I., Hryniewicz, W., Lomonaco, S., Skoczynska, A. (2018). Molecular diversity and antimicrobial susceptibility of Listeria monocytogenes isolates from invasive infections in Poland (1997-2013). Scientific Reports, 8(1), 14562. https://doi.org/10.1038/s41598-018-32574-0
Lee, S., Chen, Y., Gorski, L., Ward, T. J., Osborne, J., Kathariou, S. (2018). Listeria monocytogenes Source Distribution Analysis Indicates Regional Heterogeneity and Ecological Niche Preference among Serotype 4b Clones. MBio, 9(2), e00396-18. https://doi.org/10.1128/mBio.00396-18
Lüth, S., Halbedel, S., Rosner, B., Wilking, H., Holzer, A., Roedel, A., Dieckmann, R., Vincze, S., Prager, R., Flieger, A., Al Dahouk, S., Kleta, S. (2020). Backtracking and forward checking of human listeriosis clusters identified a multiclonal outbreak linked to Listeria monocytogenes in meat products of a single producer. Emerging Microbes & Infections, 9(1), 1600-1608. https://doi.org/10.1080/22221751.2020.1784044
Maury, M. M., Bracq-Dieye, H., Huang, L., Vales, G., Lavina, M., Thouvenot, P., Disson, O., Leclercq, A., Brisse, S., Lecuit, M. (2019). Hypervirulent Listeria monocytogenes clones' adaption to mammalian gut accounts for their association with dairy products. Nature Communications, 10(1), 2488. https://doi.org/10.1038/s41467-019-10380-0
Maury, M. M., Tsai, Y.-H., Charlier, C., Touchon, M., Chenal-Francisque, V., Leclercq, A., Criscuolo, A., Gaultier, C., Roussel, S., Brisabois, A., Disson, O., Rocha, E. P. C., Brisse, S., Lecuit, M. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nature Genetics, 48(3), 308-313. https://doi.org/10.1038/ng.3501
Montero, D., Bodero, M., Riveros, G., Lapierre, L., Gaggero, A., Vidal, R. M., Vidal, M. (2015). Molecular epidemiology and genetic diversity of Listeria monocytogenes isolates from a wide variety of ready-to-eat foods and their relationship to clinical strains from listeriosis outbreaks in Chile. Frontiers in Microbiology, 6. https://doi.org/10.3389/fmicb.2015.00384
Mota, M. I., Vázquez, S., Cornejo, C., D'Alessandro, B., Braga, V., Caetano, A., Betancor, L., Varela, G. (2020). Does Shiga Toxin-Producing Escherichia coli and Listeria monocytogenes Contribute Significantly to the Burden of Antimicrobial Resistance in Uruguay? Frontiers in Veterinary Science, 7, 583930. https://doi.org/10.3389/fvets.2020.583930
Moura, A., Tourdjman, M., Leclercq, A., Hamelin, E., Laurent, E., Fredriksen, N., Van Cauteren, D., Bracq-Dieye, H., Thouvenot, P., Vales, G., Tessaud-Rita, N., Maury, M. M., Alexandru, A., Criscuolo, A., Quevillon, E., Donguy, M.-P., Enouf, V., de Valk, H., Brisse, S., Lecuit, M. (2017). Real-Time Whole-Genome Sequencing for Surveillance of Listeria monocytogenes , France. Emerging Infectious Diseases, 23(9), 1462-1470. https://doi.org/10.3201/eid2309.170336
Pettengill, J. B., Luo, Y., Davis, S., Chen, Y., Gonzalez-Escalona, N., Ottesen, A., Rand, H., Allard, M. W., Strain, E. (2014). An evaluation of alternative methods for constructing phylogenies from whole genome sequence data: A case study with Salmonella. PeerJ, 2, e620. https://doi.org/10.7717/peerj.620
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., Ramirez, M., Carriço, J. A. (2018). chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microbial Genomics, 4(3). https://doi.org/10.1099/mgen.0.000166
Thitiananpakorn, K., Aiba, Y., Tan, X.-E., Watanabe, S., Kiga, K., Sato'o, Y., Boonsiri, T., Li, F.-Y., Sasahara, T., Taki, Y., Azam, A. H., Zhang, Y., Cui, L. (2020). Association of mprF mutations with cross-resistance to daptomycin and vancomycin in methicillin-resistant Staphylococcus aureus (MRSA). Scientific Reports, 10(1), 16107. https://doi.org/10.1038/s41598-020-73108-x
Wang, H., Luo, L., Zhang, Z., Deng, J., Wang, Y., Miao, Y., Zhang, L., Chen, X., Liu, X., Sun, S., Xiao, B., Li, Q., Ye, C. (2018). Prevalence and molecular characteristics of Listeria monocytogenes in cooked products and its comparison with isolates from listeriosis cases. Frontiers of Medicine, 12(1), 104-112. https://doi.org/10.1007/s11684-017-0593-9
Zhang, X., Niu, Y., Liu, Y., Lu, Z., Wang, D., Cui, X., Chen, Q., Ma, X. (2019). Isolation and Characterization of Clinical Listeria monocytogenes in Beijing, China, 2014-2016. Frontiers in Microbiology, 10, 981. https://doi.org/10.3389/fmicb.2019.00981
Zhou, Z., Alikhan, N.-F., Sergeant, M. J., Luhmann, N., Vaz, C., Francisco, A. P., Carriço, J. A., Achtman, M. (2018). GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Research, 28(9), 1395-1404. https://doi.org/10.1101/gr.232397.117
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114-2120. https://doi.org/10.1093/bioinformatics/btu170
Camargo, A. C., Moura, A., Avillan, J., Herman, N., McFarland, A. P., Sreevatsan, S., Call, D. R., Woodward, J. J., Lecuit, M., Nero, L. A. (2019). Whole-genome sequencing reveals Listeria monocytogenes diversity and allows identification of long term persistent strains in Brazil. Environmental Microbiology, 21(12), 4478-4487. https://doi.org/10.1111/1462-2920.14726
Chen, Y., Luo, Y., Carleton, H., Timme, R., Melka, D., Muruvanda, T., Wang, C., Kastanis, G., Katz, L. S., Turner, L., Fritzinger, A., Moore, T., Stones, R., Blankenship, J., Salter, M., Parish, M., Hammack, T. S., Evans, P. S., Tarr, C. L., � Brown, E. W. (2017). Whole Genome and Core Genome Multilocus Sequence Typing and Single Nucleotide Polymorphism Analyses of Listeria monocytogenes Isolates Associated with an Outbreak Linked to Cheese, United States, 2013. Applied and Environmental Microbiology, 83(15), e00633-17, e00633-17. https://doi.org/10.1128/AEM.00633-17
Doumith, M., Buchrieser, C., Glaser, P., Jacquet, C., Martin, P. (2004). Differentiation of the Major Listeria monocytogenes Serovars by Multiplex PCR. Journal of Clinical Microbiology, 42(8), 3819-3822. https://doi.org/10.1128/JCM.42.8.3819-3822.2004
European Centre for Disease Prevention and Control. (2016). Expert opinion on whole genome sequencing for public health surveillance: Strategy to harness whole genome sequencing to strengthen EU outbreak investigations and public health surveillance. Publications Office. https://data.europa.eu/doi/10.2900/12442
European Food Safety Authority and European Centre for Disease Prevention and Control (EFSA and ECDC). (2019). The European Union One Health 2018 Zoonoses Report. EFSA Journal, 17(12). https://doi.org/10.2903/j.efsa.2019.5926
European Food Safety Authority, European Centre for Disease Prevention and Control. (2021). The European Union One Health 2019 Zoonoses Report. EFSA Journal, 19(2). https://doi.org/10.2903/j.efsa.2021.6406
Feijao, P., Yao, H.-T., Fornika, D., Gardy, J., Hsiao, W., Chauve, C., Chindelevitch, L. (s.d.-a). MentaLiST - A fast MLST caller for large MLST schemes. Microbial Genomics, 8.
Feijao, P., Yao, H.-T., Fornika, D., Gardy, J., Hsiao, W., Chauve, C., Chindelevitch, L. (s.d.-b). MentaLiST - A fast MLST caller for large wgMLST schemes. 15.
Guidi, F., Orsini, M., Chiaverini, A., Torresi, M., Centorame, P., Acciari, V. A., Salini, R., Palombo, B., Brandi, G., Amagliani, G., Schiavano, G. F., Massacci, F. R., Fisichella, S., Domenico, M. D., Ancora, M., Pasquale, A. D., Duranti, A., Cammà, C., Pomilio, F., Blasi, G. (2021). Hypo- and Hyper-Virulent Listeria monocytogenes Clones Persisting in Two Different Food Processing Plants of Central Italy. Microorganisms, 9(2), 376. https://doi.org/10.3390/microorganisms9020376
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics, 29(8), 1072-1075. https://doi.org/10.1093/bioinformatics/btt086
Haubert, L., Kremer, F. S., da Silva, W. P. (2018). Whole-genome sequencing identification of a multidrug-resistant Listeria monocytogenes serotype 1/2a isolated from fresh mixed sausage in southern Brazil. Infection, Genetics and Evolution, 65, 127-130. https://doi.org/10.1016/j.meegid.2018.07.028
Kérouanton, A., Marault, M., Petit, L., Grout, J., Dao, T. T., Brisabois, A. (2010). Evaluation of a multiplex PCR assay as an alternative method for Listeria monocytogenes serotyping. Journal of Microbiological Methods, 80(2), 134-137. https://doi.org/10.1016/j.mimet.2009.11.008
Khademi, F., Sahebkar, A. (2019). A systematic review and meta-analysis on the prevalence of antibiotic-resistant Listeria species in food, animal and human specimens in Iran. Journal of Food Science and Technology, 56(12), 5167-5183. https://doi.org/10.1007/s13197-019-04040-w
Kuch, A., Goc, A., Belkiewicz, K., Filipello, V., Ronkiewicz, P., Golebiewska, A., Wróbel, I., Kiedrowska, M., Wasko, I., Hryniewicz, W., Lomonaco, S., Skoczynska, A. (2018). Molecular diversity and antimicrobial susceptibility of Listeria monocytogenes isolates from invasive infections in Poland (1997-2013). Scientific Reports, 8(1), 14562. https://doi.org/10.1038/s41598-018-32574-0
Lee, S., Chen, Y., Gorski, L., Ward, T. J., Osborne, J., Kathariou, S. (2018). Listeria monocytogenes Source Distribution Analysis Indicates Regional Heterogeneity and Ecological Niche Preference among Serotype 4b Clones. MBio, 9(2), e00396-18. https://doi.org/10.1128/mBio.00396-18
Lüth, S., Halbedel, S., Rosner, B., Wilking, H., Holzer, A., Roedel, A., Dieckmann, R., Vincze, S., Prager, R., Flieger, A., Al Dahouk, S., Kleta, S. (2020). Backtracking and forward checking of human listeriosis clusters identified a multiclonal outbreak linked to Listeria monocytogenes in meat products of a single producer. Emerging Microbes & Infections, 9(1), 1600-1608. https://doi.org/10.1080/22221751.2020.1784044
Maury, M. M., Bracq-Dieye, H., Huang, L., Vales, G., Lavina, M., Thouvenot, P., Disson, O., Leclercq, A., Brisse, S., Lecuit, M. (2019). Hypervirulent Listeria monocytogenes clones' adaption to mammalian gut accounts for their association with dairy products. Nature Communications, 10(1), 2488. https://doi.org/10.1038/s41467-019-10380-0
Maury, M. M., Tsai, Y.-H., Charlier, C., Touchon, M., Chenal-Francisque, V., Leclercq, A., Criscuolo, A., Gaultier, C., Roussel, S., Brisabois, A., Disson, O., Rocha, E. P. C., Brisse, S., Lecuit, M. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nature Genetics, 48(3), 308-313. https://doi.org/10.1038/ng.3501
Montero, D., Bodero, M., Riveros, G., Lapierre, L., Gaggero, A., Vidal, R. M., Vidal, M. (2015). Molecular epidemiology and genetic diversity of Listeria monocytogenes isolates from a wide variety of ready-to-eat foods and their relationship to clinical strains from listeriosis outbreaks in Chile. Frontiers in Microbiology, 6. https://doi.org/10.3389/fmicb.2015.00384
Mota, M. I., Vázquez, S., Cornejo, C., D'Alessandro, B., Braga, V., Caetano, A., Betancor, L., Varela, G. (2020). Does Shiga Toxin-Producing Escherichia coli and Listeria monocytogenes Contribute Significantly to the Burden of Antimicrobial Resistance in Uruguay? Frontiers in Veterinary Science, 7, 583930. https://doi.org/10.3389/fvets.2020.583930
Moura, A., Tourdjman, M., Leclercq, A., Hamelin, E., Laurent, E., Fredriksen, N., Van Cauteren, D., Bracq-Dieye, H., Thouvenot, P., Vales, G., Tessaud-Rita, N., Maury, M. M., Alexandru, A., Criscuolo, A., Quevillon, E., Donguy, M.-P., Enouf, V., de Valk, H., Brisse, S., Lecuit, M. (2017). Real-Time Whole-Genome Sequencing for Surveillance of Listeria monocytogenes , France. Emerging Infectious Diseases, 23(9), 1462-1470. https://doi.org/10.3201/eid2309.170336
Pettengill, J. B., Luo, Y., Davis, S., Chen, Y., Gonzalez-Escalona, N., Ottesen, A., Rand, H., Allard, M. W., Strain, E. (2014). An evaluation of alternative methods for constructing phylogenies from whole genome sequence data: A case study with Salmonella. PeerJ, 2, e620. https://doi.org/10.7717/peerj.620
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., Ramirez, M., Carriço, J. A. (2018). chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microbial Genomics, 4(3). https://doi.org/10.1099/mgen.0.000166
Thitiananpakorn, K., Aiba, Y., Tan, X.-E., Watanabe, S., Kiga, K., Sato'o, Y., Boonsiri, T., Li, F.-Y., Sasahara, T., Taki, Y., Azam, A. H., Zhang, Y., Cui, L. (2020). Association of mprF mutations with cross-resistance to daptomycin and vancomycin in methicillin-resistant Staphylococcus aureus (MRSA). Scientific Reports, 10(1), 16107. https://doi.org/10.1038/s41598-020-73108-x
Wang, H., Luo, L., Zhang, Z., Deng, J., Wang, Y., Miao, Y., Zhang, L., Chen, X., Liu, X., Sun, S., Xiao, B., Li, Q., Ye, C. (2018). Prevalence and molecular characteristics of Listeria monocytogenes in cooked products and its comparison with isolates from listeriosis cases. Frontiers of Medicine, 12(1), 104-112. https://doi.org/10.1007/s11684-017-0593-9
Zhang, X., Niu, Y., Liu, Y., Lu, Z., Wang, D., Cui, X., Chen, Q., Ma, X. (2019). Isolation and Characterization of Clinical Listeria monocytogenes in Beijing, China, 2014-2016. Frontiers in Microbiology, 10, 981. https://doi.org/10.3389/fmicb.2019.00981
Zhou, Z., Alikhan, N.-F., Sergeant, M. J., Luhmann, N., Vaz, C., Francisco, A. P., Carriço, J. A., Achtman, M. (2018). GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Research, 28(9), 1395-1404. https://doi.org/10.1101/gr.232397.117
| OPEN REVIEW - Modulo per la "revisione aperta" di questo articolo, pubblicato sul numero 124/2021 di SPVet.it |

Guidi et al., 2021 - Ceppi clinici di Listeria monocytogenes isolati nell'Umbria e nelle Marche nel biennio 2019-2020: come è cambiata la sorveglianza molecolare nell'era del Whole Genome Sequencing - Clinical strains of Listeria monocytogenes isolated in Umbria and Marche Regions (Italy) in 2019-2020: how molecular surveillance has changed in the age of Whole Genome Sequencing (SPVet.it 124/2021)

Ceppi clinici di Listeria monocytogenes isolati nell'Umbria e nelle Marche nel biennio 2019-2020: come è cambiata la sorveglianza molecolare nell'era del Whole Genome Sequencing - Clinical strains of Listeria monocytogenes isolated in Umbria and Marche Regions (Italy) in 2019-2020: how molecular surveillance has changed in the age of Whole Genome Sequencing by Guidi et al., 2021 is licensed under a Creative Commons Attribution 4.0 International License.
Permissions beyond the scope of this license may be available at http://indice.spvet.it/adv.html.